- Highlights
- Quantum mechanical restraints for proteins (article).
- phenix.magref: new tool for pixel size (magnification) refinement for cryoEM
- Phenix help chatbot available at https://phenix-online.org/chatbot
- Full list of changes: Changelog
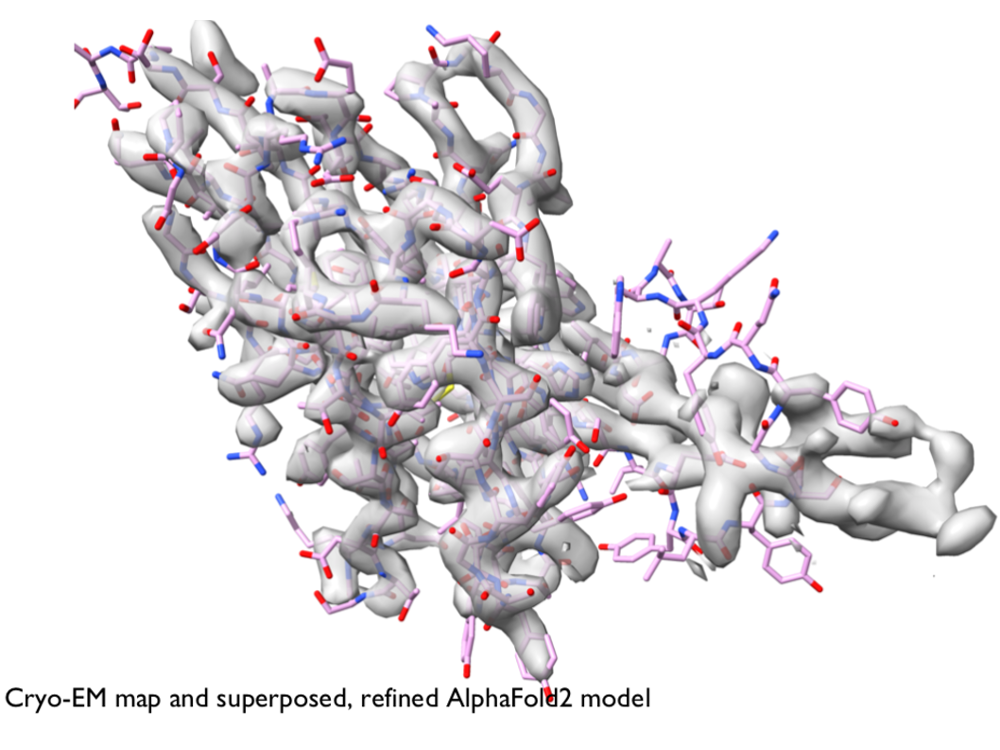
- Structure determination with AlphaFold video tutorial
- Predict a structure on the Phenix AlphaFold server video tutorial
- PredictAndBuild (Xray) video tutorial
- PredictAndBuild (cryo-EM) video tutorial
- Experimental Phasing (crystallography)
- Molecular Replacement
- Automated model building (cryo-EM and crystallography)
- Refinement (real space and reciprocal space)
- Validation

Lincoln, Nebrasca, April 30, 2026
IUCr congress in Calgary, August 10-11, 2026: link.
Other events with Phenix talks:
MCCS2026 in Madrid, Spain, March 23–27, link.
CCP4 workshop at Argonne, July 2026 link.
Recordings of past workshops
Presentations from past workshops
Citing Phenix:
Macromolecular structure determination using x-rays, neutrons and electrons: recent developments in phenix.
D. Liebschner, P. V. Afonine, M. L. Baker, G. Bunkóczi, V. B. Chen, T. I. Croll, B. Hintze, L. W. Hung, S. Jain, A. J. McCoy, N. W. Moriarty, R. D. Oeffner, B. K. Poon, M. G. Prisant, R. J. Read, J. S. Richardson, D. C. Richardson, M. D. Sammito, O. V. Sobolev, D. H. Stockwell, T. C. Terwilliger, A. G. Urzhumtsev, L. L. Videau, C. J. Williams, P. D. Adams.
Acta Crystallogr D Struct Biol
75, 861–877 (2019).
doi:10.1107/S2059798319011471.
Phenix Development, Maintenance and Distribution is Supported by:
NIH/NIGMS Program Project Grant (P01GM063210)
NIH/NIGMS R24 National Resource Grant (R24GM141254)
The Phenix Industrial Consortium