| Python-based Hierarchical ENvironment for Integrated Xtallography |
| Documentation Home |
Hybrid Substructure Search
HySS overviewThe HySS (Hybrid Substructure Search) submodule of the Phenix package is a highly-automated procedure for the location of anomalous scatterers in macromolecular structures. HySS starts with the automatic detection of the reflection file format and analyses all available datasets in a given reflection file to decide which of these is best suited for solving the structure. The search parameters are automatically adjusted based on the available data and the number of expected sites given by the user. The search method is a systematic multi-trial procedure employing The end result is a consensus model which is exported in a variety of file formats suitable for frequently used phasing and density modification packages. Links: The core search procedure is applicable to both anomalous diffraction and isomorphous replacement problems. However, currently the command line interface is limited to work with anomalous diffraction data or externally preprocessed difference data. References: To contact us send email to help@phenix-online.org or bugs@phenix-online.org. HySS examplesThe only input file required for running HySS is a file with the reflection data. HySS reads the following formats directly:
nsf_d2_peak.scaThe CCI Apps binary bundles include a scalepack file with anomalous peak data for the structure with the PDB access code 1NSF (courtesy of A.T. Brunger). To find the 8 selenium sites enter: phenix.hyss nsf_d2_peak.sca 8 seThis leads to: Reading reflection file: nsf_d2_peak.sca Space group found in file: P 6 Is this the correct space group? [Y/N]:HySS prompts for a confirmation of the space group because space group P6 is often used as a placeholder during data reduction. If the space group symbol found in the reflection file is not correct it can be changed. However, in this case the symbol is correct. At the prompt enter Y to continue. Alternatively, the interactive prompt can be avoided by using the --space_group option: phenix.hyss nsf_d2_peak.sca 8 se --space_group=p6HySS will quickly print a few screen-pages with information about the data (e.g. the magnitude of the anomalous signal) and the many search parameters. The most interesting output is produced after this point: Entering search loop: p = peaklist index in Patterson map f = peaklist index in two-site translation function ess = score after extrapolation scan r = number of dual-space recycling cycles score = final score p=000 f=000 ess=0.364 (cc) r=015 score=0.479 (cc) [ best score: 0.479 (cc) ] p=000 f=001 ess=0.310 (cc) r=015 score=0.477 (cc) [ best score: 0.479 (cc) 0.477 (cc) ] Number of matching sites of top 2 structures: 11 p=000 f=002 ess=0.166 (cc) r=015 score=0.479 (cc) [ best score: 0.479 (cc) 0.479 (cc) 0.477 (cc) ] Number of matching sites of top 2 structures: 11 Number of matching sites of top 3 structures: 11It will take a few seconds for each line starting with p= to appear. Each of these lines summarizes the result of one trial consisting of an evaluation of the Patterson function, two fast translation functions, and 15 cycles of dual-space recycling. The important number to watch is the final correlation. In the first three trials HySS finds three substructure models with promisingly high correlations. These models are compared, taking allowed origin shifts and the hand ambiguity into account. The three models have more than 2/3 of the expected number of sites in common. Therefore HySS decides that the search is complete and prints a summary of the matching sites:
Top 3 scores:
p=000 f=000 ess=0.364 (cc) r=015 score=0.479 (cc)
p=000 f=001 ess=0.310 (cc) r=015 score=0.477 (cc)
p=000 f=002 ess=0.166 (cc) r=015 score=0.479 (cc)
Match summary:
Operator:
rotation: {{-1.0, 0.0, 0.0}, {0.0, -1.0, 0.0}, {0.0, 0.0, -1.0}}
translation: (-9.6289517721653785e-38, 0.0, 0.091526465343537006)
rms coordinate differences: 0.06
Pairs: 11
site001 site001 0.018
site002 site002 0.056
site003 site003 0.033
site004 site004 0.026
site005 site005 0.050
site006 site006 0.103
site007 site007 0.040
site008 site008 0.063
site009 site010 0.067
site010 site009 0.120
site011 site011 0.029
Singles model 1: 0
Singles model 2: 0
The matching sites are used to build a consensus model. The
coordinates and occupancies are quickly refined using a quasi-Newton
minimizer:
Minimizing consensus model (11 sites). Truncating consensus model to expected number of sites. Minimizing consensus model (8 sites). Correlation coefficient for consensus model (8 sites): 0.483The refined sites are sorted by occupancy in descending order. The model is truncated to the expected number of sites and refined again. After printing detailed timing information (not shown) the output ends with: Storing all substructures found: nsf_d2_peak_hyss_models.pickle Storing consensus model: nsf_d2_peak_hyss_consensus_model.pickle Writing consensus model as PDB file: nsf_d2_peak_hyss_consensus_model.pdb Writing consensus model as CNS SDB file: nsf_d2_peak_hyss_consensus_model.sdb Writing consensus model as SOLVE xyz records: nsf_d2_peak_hyss_consensus_model.xyz The fractional coordinates may also be useful in other programs. Total CPU time: 49.60 secondsThe resulting coordinate files can be used for phasing and density modification with other programs. gere_MAD.mtzThe CCP4 distribution includes a four-wavelength MAD dataset in the tutorial directory. To find the 12 selenium sites with HySS enter: phenix.hyss $CEXAM/tutorial2000/data/gere_MAD.mtz 12 seHySS automatically picks the wavelength with the strongest anomalous signal and finishes after about 34 seconds (2.8GHz Pentium 4 Linux), writing out the 12 (or sometimes only 11) sites in the various file formats. mbp.hklThe CNS tutorial includes data from a MAD experiment with Ytterbium as the anomalous scatterer. CNS reflection files do not contain information about the unit cell and space group. However, HySS is able to extract this information from other files, e.g. other reflection files, CNS files, SOLVE files, PDB files or SHELX files. For example: phenix.hyss $CNS_SOLVE/doc/html/tutorial/data/mbp/mbp.hkl 4 yb --symmetry $CNS_SOLVE/doc/html/tutorial/data/mbp/defHySS reads the reflection data from the mbp.hkl file. The --symmetry options instructs HySS to scan the def file for unit cell parameters and a space group symbol. HySS finishes after about 26 seconds (2.8GHz Pentium 4 Linux). Graphical interfaceThe HySS GUI is listed in the "Experimental phasing" category of the main PHENIX GUI. Most options are shown in the main window, but only the fields highlighted below are mandatory. The data labels will be selected automatically if the reflections file contains anomalous arrays, and any symmetry information present in the file will be loaded in the unit cell and space group fields. 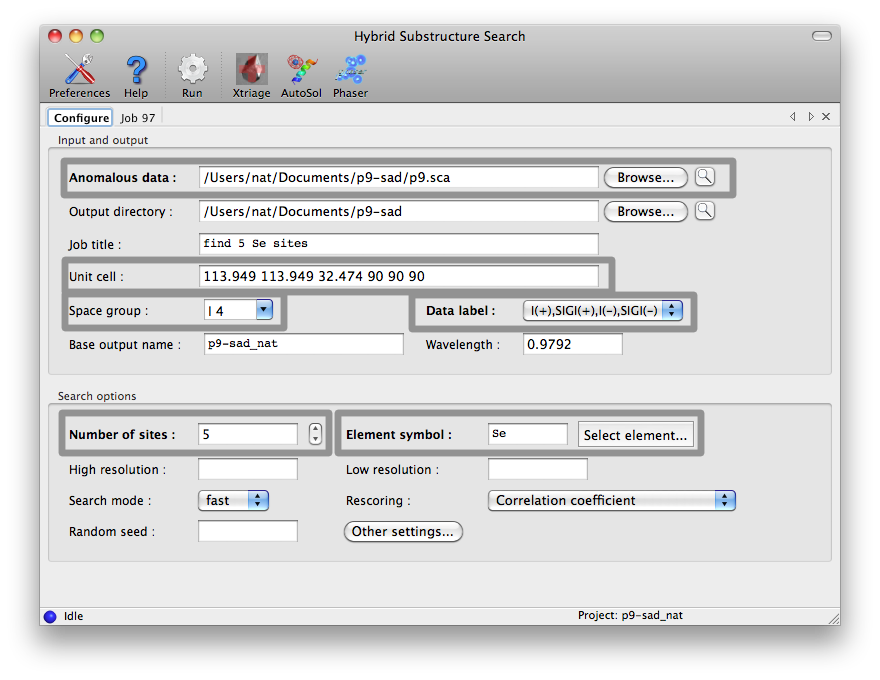 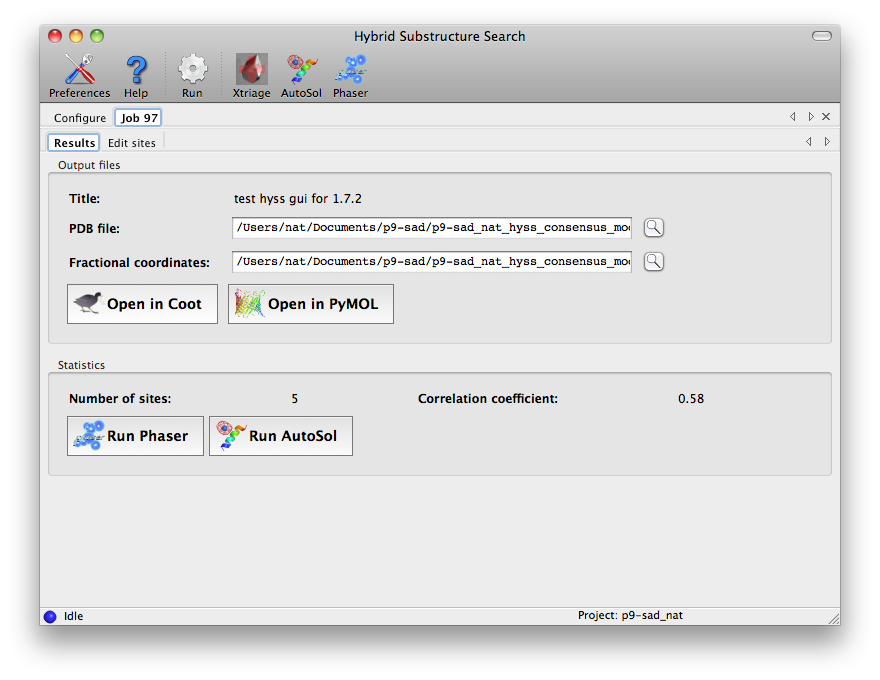 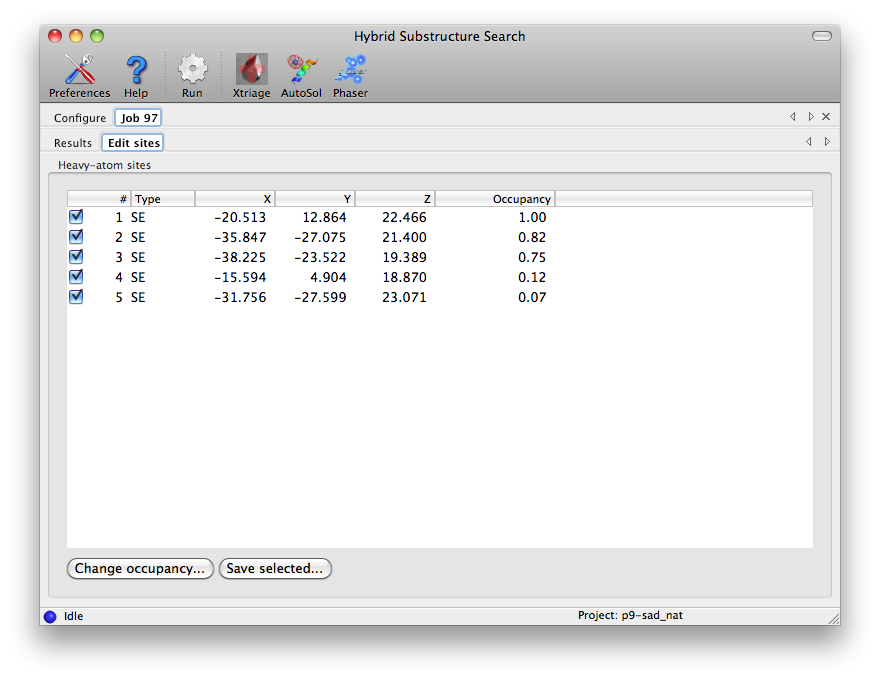 Command line optionsEnter phenix.hyss without arguments to obtain a list of the available command line options:
Command line arguments:
usage: phenix.hyss [options] reflection_file n_sites element_symbol
options:
-h, --help show this help message and exit
--unit_cell=10,10,20,90,90,120|FILENAME
External unit cell parameters
--space_group=P212121|FILENAME
External space group symbol
--symmetry=FILENAME External file with symmetry information
--chunk=n,i Number of chunks for parallel execution and index for
one process
--search=fast|full Search mode
--resolution=FLOAT High resolution limit (minimum d-spacing, d_min)
--low_resolution=FLOAT
Low resolution limit (maximum d-spacing, d_max)
--site_min_distance=FLOAT
Minimum distance between substructure sites (default:
3.5)
--site_min_distance_sym_equiv=FLOAT
Minimum distance between symmetrically-equivalent
substructure sites (overrides --site_min_distance)
--site_min_cross_distance=FLOAT
Minimum distance between substructure sites not
related by symmetry (overrides --site_min_distance)
--molecular_weight=FLOAT
Molecular weight
--solvent_content=FLOAT
Solvent content (default: 0.55)
--random_seed=INT Seed for random number generator
--real_space_squaring
Use real space squaring (as opposed to the tangent
formula)
--data_label=STRING Substring of reflection data label
--rescore=correlation|phaser-refine|phaser-complete
Select rescoring protocol (default: correlation).
Phaser-based protocols are more computationally
intensive, but slightly more discriminative towards
correct solutions, and may identify solutions if
default protocol is not conclusive
--extrapolation=fast_nv1995|phaser-map
Select extrapolation protocol (default:
fast_nv1995). Fast_nv1995 uses a fast translation
function to find atoms in the difference Patterson
function, while phaser-map calculates a SAD LLG map
and locates peaks.
See also:
http://www.phenix-online.org/download/documentation/cci_apps/hyss/
Example: phenix.hyss w1.sca 66 Se
The --data_label, --resolution and --low_resolution options
can be used to override the automatic selection of the reflection data
and the resolution range. For example, one may enter the following
command with the goal to instruct HySS to use the peak data in the
gere_MAD.mtz file (instead of the inflection point data), and to
set the high resolution limit to 5 Angstrom:
phenix.hyss gere_MAD.mtz 12 se --data_label=peak --resolution=5Output: Command line arguments: gere_MAD.mtz 12 se --data_label=peak --resolution=5 Reading reflection file: gere_MAD.mtz Ambiguous --data_label=peak Possible choices: 5: gere_MAD.mtz:FSEpeak,SIGFSEpeak,DSEpeak,SIGDSEpeak,merged 6: gere_MAD.mtz:F(+)SEpeak,SIGF(+)SEpeak,F(-)SEpeak,SIGF(-)SEpeak Please specify an unambiguous substring of the target label. Sorry: Please try again.That's a good first try but if --data_label=peak turns out to be ambiguous HySS will ask for more information. Second try: phenix.hyss gere_MAD.mtz 12 se --data_label="F(+)SEpeak" --resolution=5Now HySS will actually perform the search. Typically the search finishes in less than 10 seconds finding 8-12 sites, depending on the random number generator (which is seeded with the current time unless the --random_seed option is used). The --site_min_distance, --site_min_distance_sym_equiv, and --site_min_cross_distance options are available to override the default minimum distance of 3.5 Angstroms between substructure sites. The --real_space_squaring option can be useful for large structures with high-resolution data. In this case the large number of triplets generated for the reciprocal-space direct methods procedure (i.e. the tangent formula) may lead to excessive memory allocation. By default HySS switches to real-space direct methods (i.e. E-map squaring) if it searches for more than 100 sites. If this limit is too high given the available memory use the --real_space_squaring option. For substructures with a large number of sites it is in our experience not critical to employ reciprocal-space direct methods. If the --molecular_weight and --solvent_content options are used HySS will help in determining the number of substructures sites in the unit cell, interpreting the number of sites specified on the command line as number of sites per molecule. For example: phenix.hyss gere_MAD.mtz 2 se --molecular_weight=8000 --solvent_content=0.70This is telling HySS that we have a molecule with a molecular weight of 8 kD, a crystal with an estimated solvent content of 70%, and that we expect to find 2 Se sites per molecule. The HySS output will now show the following: #---------------------------------------------------------------------------# | Formula for calculating the number of molecules given a molecular weight. | |---------------------------------------------------------------------------| | n_mol = ((1.0-solvent_content)*v_cell)/(molecular_weight*n_sym*.783) | #---------------------------------------------------------------------------# Number of molecules: 6 Number of sites: 12 Values used in calculation: Solvent content: 0.70 Unit cell volume: 476839 Molecular weight: 8000.00 Number of symmetry operators: 4HySS will go on searching for 12 sites. If things go wrongIf the HySS consensus model does not lead to an interpretable electron density map please try the --search full option: phenix.hyss your_file.sca 100 se --search fullThis disables the automatic termination detection and the run will in general take considerably longer. If the full search leads to a better consensus model please let us know because we will want to improve the automatic termination detection. Another possibility is to override the automatic determination of the high-resolution limit with the --resolution option. In some cases the resolution limit is very critical. Truncating the high-resolution limit of the data can sometimes lead to a successful search, as more reflections with a weak anomalous signal are excluded. Enabling a phaser-based rescoring protocol can also help (--rescore=phaser-complete is recommended). It is less affected by suboptimal resolution cutoffs and also provides more discrimination with noisy data. Switching on the phaser-map extrapolation protocol is also worthwhile, since it increases success rate and is only a small runtime overhead compared to phaser-based rescoring. If there is no consensus model at the end of a HySS run please try alternative programs. For example, run SHELXD with the .ins and .hkl files that are automatically generated by HySS: Writing anomalous differences as SHELX HKLF file: mbp_anom_diffs.hkl Writing SHELXD ins file: mbp_anom_diffs.insIf HySS does not produce a consensus model even though it is possible to solve the substructure with other programs we would like to investigate. Please send email to bugs@phenix-online.org. Auxiliary programsphenix.emmaEMMA stands for Euclidean Model Matching which allows two sets of coordinates to be superimposed as best as possible given symmetry and origin choices. See the phenix.emma documentation for more details. phenix.xtriageThe phenix.xtriage program performs an extensive suite of tests to assess the quality of a data set. It is a good idea to always run this program before substructure location or any other steps of structure solution. See the phenix.xtriage documentation for more details. phenix.reflection_statisticsComparision between multiple datasets is available using the phenix.reflection_statistics command. See the phenix.reflection_statistics documentation for more details. | |