Merging reflections with the Phenix GUI
- Dealing with R-free flags
- Notes on usage
Phenix includes a simple program for combining reflection data from
different files and manipulating R-free flags, somewhat similar to the
programs CAD and FREERFLAG in the CCP4 suite. Any combination of data in
the form of Miller arrays (any set of values indexed by h,k,l) may be merged
into a single file, with the limitation that the crystal lattices
specified in each file must be compatible. All input formats readable by
Phenix are supported, but only MTZ files are written. Output is limited to
25 different columns, which usually corresponds to significantly fewer
Miller arrays (for example, experimental phases or Hendrickson-Lattman
coefficients consist of four columns).
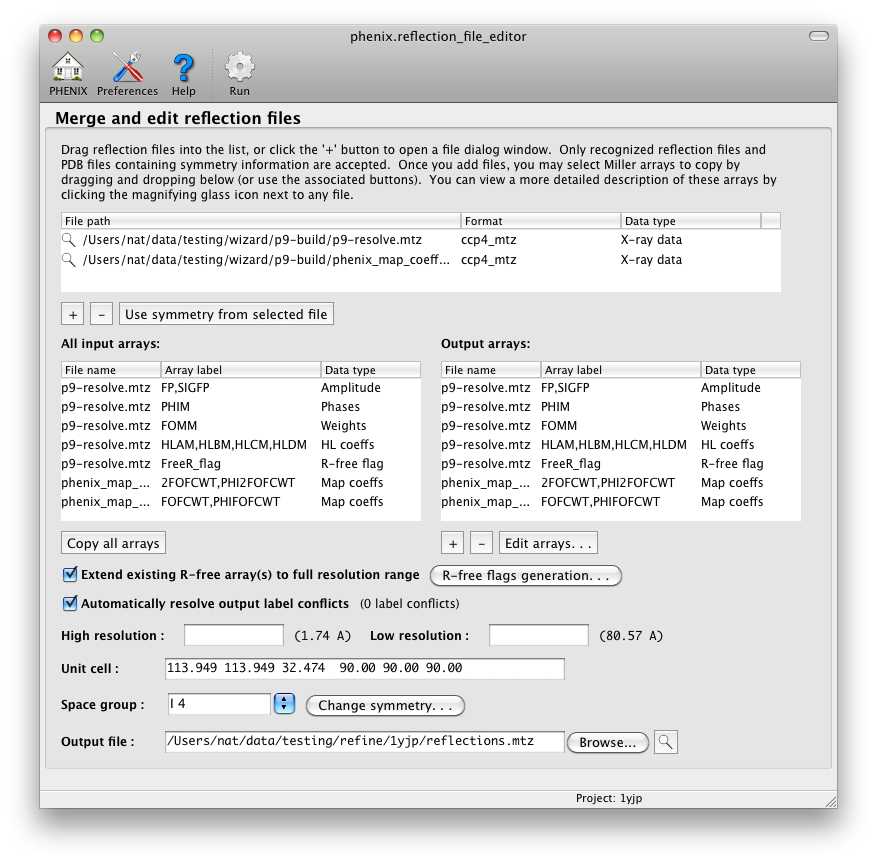
To add an input file, click the "+" button below the top list or simply drag
files from the desktop into the list. Available Miller arrays will
automatically be loaded into the input list, and the crystal symmetry (if
present in the file(s)) will be updated. Select arrays by drag-and-drop
into the output list or using the buttons below. If different arrays
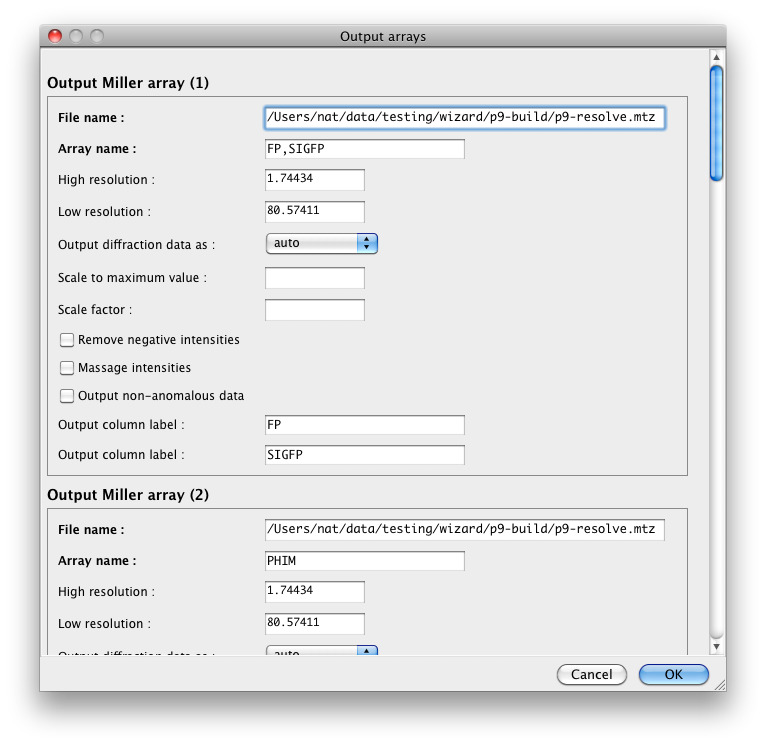
have the same column label this will be noted in the window. You can edit
the output labels and resolution limits for individual arrays by clicking
the "Edit arrays. . ." button. You can also manipulate the output data
by merging anomalous data or converting to and from intensities and amplitudes.
Dealing with R-free flags
If an R-free set is not present in the output file you may create one by
clicking the "R-free generation" button. The default behavior is to flag
5% of reflections up to a maximum of 2000. Currently the assignment of flags
is completely random across the entire resolution range, but will default to
using the highest possible lattice symmetry initially and expanding to the
actual symmetry. (In other words, if the space group is P4, the R-free set
will be generated in P422 and expanded to P4.) Alternately, you may pick
the test set in thin resolution shells, which helps avoid bias due to
non-crystallographic symmetry. (Since the test set will not be evenly
distributed across the entire resolution range, don't use this option if
NCS is not present.)
An existing R-free set may also be extended to the entire resolution range of
the output file, e.g. when switching to a higher-resolution dataset for a
partially refined structure. This will also use the highest possible lattice
symmetry, and will automatically determine the test set size. If you
previously assigned the test set in thin shells using DATAMAN or SFTOOLS,
the program will attempt to detect this and issue a warning.
Phenix uses the CNS/XPLOR convention for R-free flags, where 1 marks the test
set and 0 the "working" set. By default, all R-free arrays will be written
in this format as well, regardless of source; if you generated R-free flags in
CCP4, which assigns random numbers (usually 0 through 19) and uses 0 to mark
the test set, they will be converted first. If you prefer to preserve the
original integer values, the R-free flags dialog includes an option to do
this; however, this is incompatible with extending an existing set.
Alternately, you may choose to output all R-free flags in the CCP4 format
even if extending existing flags, but this will renumber all flags not in
the test set. (It also assumes that the test set size is approximately 5% of
the total number of reflections.)
Notes on usage
- The editor works best when all input files are in MTZ format; other
formats should work but have not been tested as thoroughly.
- Output labels will be guessed automatically, but this breaks down when
merging anomalous data, which halves the number of output arrays (and
makes the trailing (+) and (-) unnecessary). This will be checked at
runtime.
- Some data processing programs output the columns F SIGF DANO SIGDANO, which
if they occur sequentially in an MTZ file will be treated as a single data
array. Internally, these are converted to anomalous data (i.e. F(+)
SIGF(+) F(-) SIGF(-)), instead of keeping the original non-anomalous
amplitudes and separate anomalous differences. We recommend against using
these data in PHENIX, as they are less reliable than the original anomalous
amplitudes or intensities (and are not otherwise used here).
- The editor will not otherwise attempt to impose sensible label names,
except when converting between intensities and amplitudes.
- All input files must either have the same symmetry (allowing for slight
differences in unit cell dimensions and screw axes) or leave it undefined
(e.g. CNS format). To mix arrays from files with different symmetry,
first convert each to the desired final symmetry, then combine them.
- All output arrays must also have the same symmetry, including isomorphous
unit cells. (You may, however, disable the check for isomorphism.)
|